Our research tackles some fundamental evolutionary questions such as speciation, adaptation, and introgression. In particular, our work focuses on integrating experimental and computational approaches to study adaptive introgression in model and non-model organisms. Our research interests also include investigating anti-predator adaptation such as mimicry, aposematic coloration, and camouflage in insects.
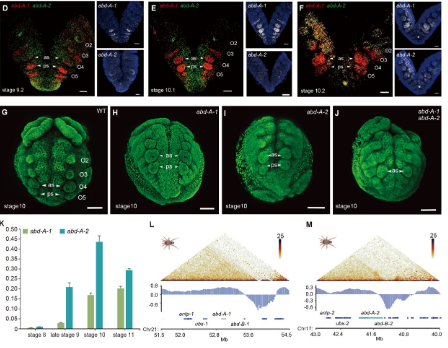
An ancient genome duplication event drives the development and evolution of spinnerets in spiders
Key appendage innovations have driven the origin and expansion of arthropods, such as spinnerets enabling spiders to occupy three-dimensional space and diversify into more than 53,000 species. Here, we investigate the genetic basis of spinneret emergence in spiders by examining the complex history and functional importance of arachnid genome evolution. Using chromosome-scale genomes from newly sequenced spiders and the whip scorpion, we integrate evidence from macrosynteny and phylogenetic analyses to provide further strong support for a whole-genome duplication (WGD) event that occurred during early Arachnopulmonata evolution. Following this event, the abdominal-A gene pair not only exhibits functional divergence but also jointly facilitates the emergence of spinnerets. Furthermore, we integrated single-cell transcriptomic analyses and functional validation to confirm that the dachshund-1 gene also regulates spinneret development. The network of duplicated gene pairs may form a cornerstone in the origin and evolution of key morphological traits, revealing that the long-term effects of ancient WGDs on innovation and diversification also occurred in arthropods.
link: https://doi.org/10.1126/sciadv.adw2173
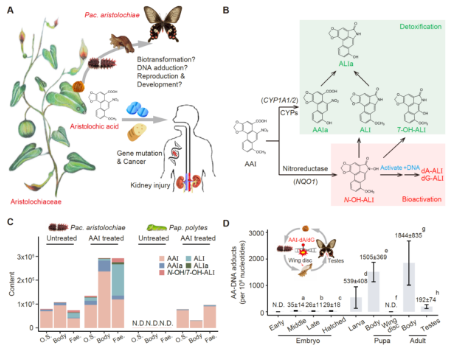
Mechanisms of Aristolochic Acid Resistance Insights
Aristolochic acids (AAs) are natural compounds found in Aristolochiaceae plants, to which humans are frequently exposed through environmental and medicinal sources. AAs are highly nephrotoxic and carcinogenic, mediated by oxidative stress and bioactivation-induced DNA damage and mutagenicity. Nevertheless, some Lepidoptera, including Pachliopta aristolochiae, feed exclusively on Aristolochiaceae and sequester AAs as a chemical defense. This is uncommon in nature and it is not yet fully understood how these insects avoid the lethal effects of AAs. To address this question, we investigate Pac. aristolochiae’s AA-resistance mechanisms by employing metabolic analyses, multiomics analyses, in situ imaging and more. Our findings indicate that AAs may be detoxified through biotransformation and a robust antioxidant system, involving candidate genes such as 15-oxo-prostaglandin 13-reductases (PGRs), cytochrome P450s, and catalases. Unexpectedly, DNA adducts, the covalent binding products from activated AA intermediates, are detected across most life stages of Pac. aristolochiae, revealing that Pac. aristolochiae can maintain genomic integrity despite a substantial burden (reaching over 1800 AA-DNA adducts per 108 nucleotides in adults, approximately 1 adduct per 55 000 nucleotides). Our study highlights insect AA tolerance as a means to discover human protective mechanisms, thereby suggesting new avenues for preventing AA-related diseases.
link: https://doi.org/10.1002/advs.202518072
link: https://doi.org/10.1002/advs.202518072
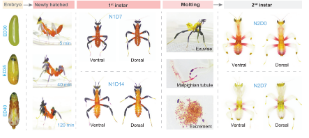
The pigment transporter Redboy confers programmed body colour transition in orchid mantises
Programmed phenotypic transition is prevalent throughout the tree of life, yet the concrete mechanisms that underpin this phenomenon are poorly understood. The orchid mantis (Hymenopus coronatus, Mantodea) is a model study system for programmed body colour transitions that displays a prominent black-red body colour in first-instar nymphs, then switches to a flowery white body colour in later-instar nymphs. Here we reveal that this body colour transition is achieved by the simultaneous excretion of decarboxylated-xanthommatin (red pigment) and the accumulation of uric acid (white pigment) in the epidermis during the first moult. This change in pigmentation is associated with a novel subtype of ABCG pigment transporter that we call ‘Redboy’ in Polyneoptera, which is upregulated by insect steroid hormone (ecdysone) during the first moult of orchid mantises. RNAi assay and pigment analyses show that Redboy functions together with the co-transporter White, exporting red pigments from and concurrently importing white pigments into the epidermal cells. Spectral reflectance analyses and predation experiments reveal that Redboy-conferred programmed body colour transition enhances predator avoidance during the first instar, and both prey attraction and predator avoidance in later instars. Our findings clarify how gene family evolution and hormone regulation coordinate programmed phenotypic transition and promote ecological adaptation in orchid mantises.
link: https://www.nature.com/articles/s41559-025-02737-0
The diversification of butterfly wing patterns: Progress and prospects
Butterfly wings display rich phenotypic diversity and are associated with complex biological functions, thus serving as an important evolutionary system to address the genetic basis and evolution of phenotypic diversification. We review recent butterfly studies that revealed complex functions underlying diversified wing patterns and describe the genetic and environmental factors involved in wing pattern determinations. These factors lead to inter-specific divergence, genetic polymorphism, and phenotypic plasticity, which in many cases are decided by several key genes. We also summarize the research advances on gene co-option as an important origin of functional complexity and evolutionary novelty. These findings reveal a pattern of evolutionary innovation within a constrained developmental framework during butterfly wing morphogenesis, but further research is required to gain a systematic and comprehensive understanding.
link: https://linkinghub.elsevier.com/retrieve/pii/S2214-5745(23)00134-7
link: https://linkinghub.elsevier.com/retrieve/pii/S2214-5745(23)00134-7

Potential and progress of studying mountain biodiversity by means of butterfly genetics and genomics
Mountains are rich in biodiversity, and butterflies are species-rich and have a good ecological and evolutionary research foundation. This review addresses the potential and progress of studying mountain biodiversity using butterflies as a model. We discuss the uniqueness of mountain ecosystems, factors influencing the distribution of mountain butterflies, representative genetic and evolutionary models in butterfly research, and evolutionary studies of mountain biodiversity involving butterfly genetics and genomics. Finally, we demonstrate the necessity of studying mountain butterflies and propose future perspectives. This review provides insights for studying the biodiversity of mountain butterflies as well as a summary of research methods for reference.
link: https://www.sciencedirect.com/science/article/pii/S1673852723001364

Inferring historical introgression with deep learning
Resolving phylogenetic relationships among taxa remains a challenge in the era of big data due to the presence of genetic admixture in a wide range of organisms. Rapidly developing sequencing technologies and statistical tests enable evolutionary relationships to be disentangled at a genome-wide level, yet many of these tests are computationally intensive and rely on phased genotypes, large sample sizes, restricted phylogenetic topologies, or hypothesis testing. To overcome these difficulties, we developed a deep learning-based approach, named ERICA, for inferring genome-wide evolutionary relationships and local introgressed regions from sequence data. ERICA accepts sequence alignments of both population genomic data and multiple genome assemblies, and efficiently identifies discordant genealogy patterns and exchanged regions across genomes when compared with other methods. We further tested ERICA using real population genomic data from Heliconius butterflies that have undergone adaptive radiation and frequent hybridization. Finally, we applied ERICA to characterize hybridization and introgression in wild and cultivated rice, revealing the important role of introgression in rice domestication and adaptation. Taken together, our findings demonstrate that ERICA provides an effective method for teasing apart evolutionary relationships using whole genome data, which can ultimately facilitate evolutionary studies on hybridization and introgression.
link: https://doi.org/10.1093/sysbio/syad033

Imperfect ant mimicry contributes to local adaptation in a jumping spider
Putative ant mimicry is a remarkable example of an evolutionary strategy that can be well integrated into the framework of natural selection and adaptation. However, challenges remain in understanding imperfect ant mimicry. Here, we combine trait quantification and behavioral assays to investigate imperfect ant mimicry in the jumping spider Siler collingwoodi. We performed trajectory analysis and gait analysis to show that the locomotor characters of S. collingwoodi generally resemble those of the putative ant models, supporting the multiple models hypothesis. We then performed background-matching analysis, revealing that body coloration may be involved in background camouflage. We further carried out antipredation assays and found that S. collingwoodi had a significantly lower risk of predation than nonmimetic salticids, suggesting an overall protective effect of Batesian mimicry. Our findings quantitatively demonstrate a combination of mimicry and camouflage in S. collingwoodi and thus highlight the significance of a complex phenomenon driven by natural selection.
link: https://www.cell.com/iscience/fulltext/S2589-0042(23)00824-6

The evolutionary mechanism of speciation in butterflies
Chemosensing plays a fundamental role in the life history of these groups of butterflies and in the establishment of reproductive isolation. However, chemical communication involves synergistic sensory and accessory functions, and it remains challenging to investigate the molecular mechanisms underlying behavioral differences. Neotropical Heliconius butterflies are well known for Müllerian mimicry, intricate behaviors, and multiple instances of radiation, which are ideal system for speciation. Here, we examine the gene expression profiles and genomic divergence of three sensory tissues (antennae, legs, and mouth parts) between sexes (females and males) and life stages (different adult stages) in two hybridizing butterflies, Heliconius melpomene and Heliconius cydno. We revealed the evolution of chemosensing in incipient speciation of butterflies between sexes and life stage.
Link: https://doi.org/10.1093/molbev/msac225
Link: https://doi.org/10.1093/molbev/msac225

The evolution and diversification of oakleaf butterflies
Oakleaf butterflies in the genus Kallima have a polymorphic wing phenotype enabling these insects to masquerade as dead leaves. This iconic example of protective resemblance provides an interesting evolutionary paradigm that can be employed to study biodiversity. We integrated multi-omic data analyses and functional validation to infer Kallima’s evolutionary history and investigate the genetic basis of their variable leaf wing patterns. We find that Kallima butterflies diversified in the eastern Himalayas and dispersed to East and Southeast Asia. Moreover, we find that leaf wing polymorphism is controlled by the wing patterning gene, cortex, which has been maintained in Kallima by long-term balancing selection. Our results provide macroevolutionary and microevolutuoanry insights into a model species originating from a mountain ecosystem.
Link:https://www.sciencedirect.com/science/article/abs/pii/S0092867422007942
Link:https://www.sciencedirect.com/science/article/abs/pii/S0092867422007942

A widely diverged locus among Heliconius butterflies may be linked to locomotor adaptation
Heliconius butterflies have undergone adaptive radiation and therefore serve as an excellent system for exploring the continuum of speciation and adaptive evolution. However, there is a long-lasting paradox between their convergent mimetic wing patterns and rapid divergence in speciation. Here, we characterize a locus that consistently displays high divergence among Heliconius butterflies and acts as an introgression hotspot. We further show that this locus contains multiple genes related to locomotion and conserved in Lepidoptera. In light of these findings, we consider that locomotion traits may be under selection, and if these are heritable traits that are selected for, then they might act as species barriers.
Link:https://advances.sciencemag.org/content/7/32/eabh2340
Link:https://advances.sciencemag.org/content/7/32/eabh2340
